Qu’est-ce que la validation GxP ?
La validation GxP repose sur un ensemble de bonnes pratiques destinées à garantir la qualité, la sécurité et la conformité des activités réglementées.
Que signifie GxP ?
Les GxP (« Good x Practice ») désignent un ensemble de réglementations, lignes directrices et bonnes pratiques qualité utilisées dans les industries réglementées. Elles couvrent ainsi plusieurs référentiels de bonnes pratiques, les principaux étant :
- GMP (Good Manufacturing Practice) – production
- GLP (Good Laboratory Practice) – laboratoire
- GCP (Good Clinical Practice) – essais cliniques
Pourquoi ces règles existent-elles ?
Les GxP existent pour garantir plusieurs objectifs :
- la sécurité du patient en évitant qu’un médicament ou dispositif médical soit :
- contaminé ;
- inefficace ;
- mal fabriqué ;
- ou dangereux.
- la qualité et la fiabilité des produits. Les GxP imposent des procédures standardisées, une documentation contrôlée, des contrôles qualité et une traçabilité complète. L’objectif ici est de produire de manière constante un produit conforme.
- l’intégrité et la traçabilité des données. En effet, les autorités réglementaires exigent que les données soient :
- exactes ;
- sécurisées ;
- auditables ;
- non falsifiables.
C’est particulièrement important pour les résultats de laboratoire, les essais cliniques, les dossiers de fabrication et la signature électronique. Nous reviendrons sur ce point ensuite.
- la conformité règlementaire lors d’audits, d’inspections d’organismes de réglementation (FDA/EMA/ANSM), de certifications. En cas de non-respect, les entreprises sont exposées à des observations réglementaires, des rappels produits, voire des suspensions de production. Cela peut même aller jusqu’à des sanctions financières.
Pourquoi les systèmes informatiques sont-ils concernés par la validation GxP ?
Dans les industries réglementées, les systèmes IT sont fortement liés aux exigences GxP car ils gèrent des données et des processus critiques ayant un impact sur les garanties qu’offrent les GxP (pour rappel : la sécurité des patients, la qualité et la fiabilité des produits, l’intégrité et la traçabilité des données et la conformité réglementaire). Cela signifie qu’il faut démontrer que le système fonctionne comme prévu et protège l’intégrité des données.
La validation GxP vient garantir que tout système informatisé utilisé dans un contexte réglementé est fiable, traçable, sécurisé et conforme aux exigences réglementaires.
C’est notamment le cas de certains ERP de production, des systèmes de gestion documentaire (GED), des logiciels qualité (QMS), des systèmes de laboratoire (LIMS) ou encore des outils de Contract Lifecycle Management (CLM), lorsque leur usage intervient dans un processus réglementé.
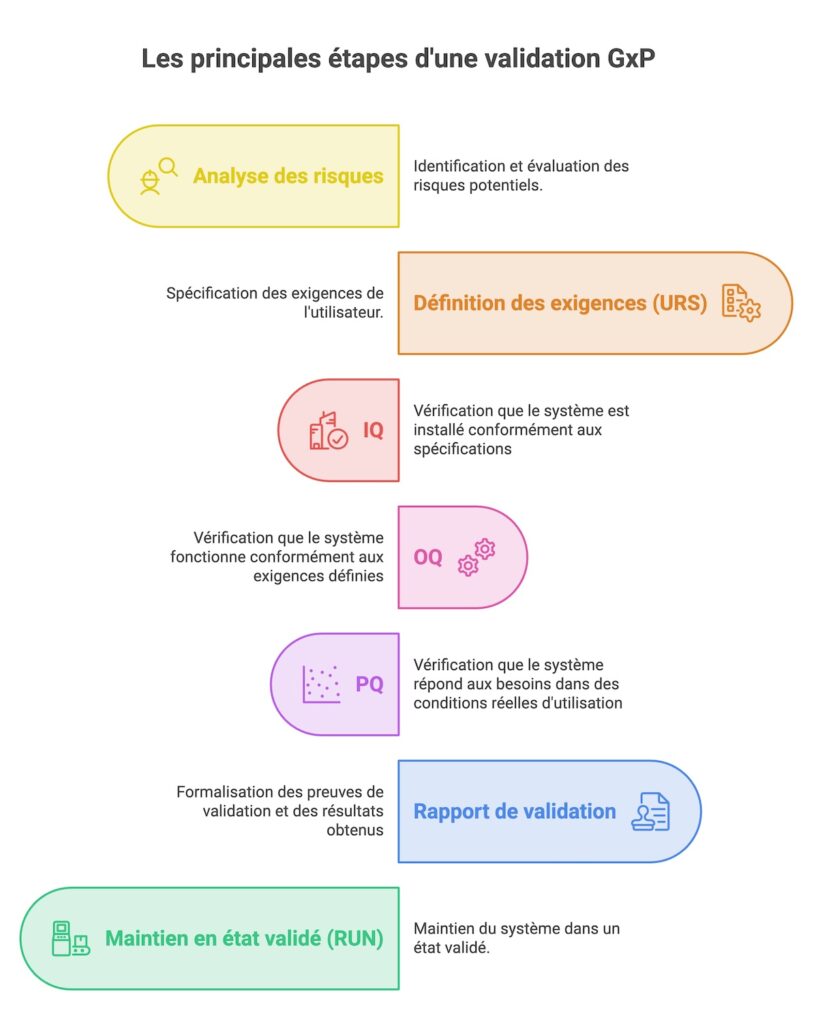
Les grandes étapes de la validation GxP
1. Définir le plan de validation
La validation GxP se déroule en plusieurs étapes. La première consiste à établir un plan de validation qui définit la stratégie globale de la démarche. Ce document de référence précise notamment le périmètre du système, l’approche fondée sur les risques, les rôles et responsabilités, les livrables attendus ainsi que le planning du projet
2. Réaliser une analyse de risques
Ensuite, il faut évaluer les risques. L’objectif de cette démarche est de prioriser les efforts de validation en concentrant les ressources sur les fonctions les plus critiques. Cela passe par l’identification des fonctionnalités ayant un impact sur la sécurité des patients, la qualité du produit ou l’intégrité des données, puis par une analyse des risques fondée sur leur gravité, leur occurrence et leur détectabilité.
Cette évaluation permet ensuite de définir les contrôles et niveaux de validation nécessaires, selon une approche proportionnée conforme aux principes GxP. Par exemple, une fonctionnalité permettant de signer électroniquement un Quality Agreement sera considérée comme plus critique qu’une simple fonction de consultation, car elle peut avoir un impact direct sur la conformité réglementaire
3. Formaliser les exigences (URS)
L’étape suivante consiste à formaliser les besoins métier et les exigences fonctionnelles et réglementaires dans un document appelé URS (User Requirements Specification). Chaque exigence doit être rédigée de manière claire, testable et traçable afin de pouvoir être vérifiée lors des phases de qualification.
Pour garantir cette traçabilité, les URS suivent généralement plusieurs bonnes pratiques :
- une numérotation unique (URS-001, URS-002…) ;
- un versioning maîtrisé ;
- une approbation conjointe des équipes métier, Qualité (QA) et IT ;
- ainsi qu’une rédaction simple, précise et cohérente avec l’analyse de risques.
Exemple : une exigence peut préciser que « le système doit enregistrer toute modification d’un contrat en conservant l’identité de l’utilisateur, la date et l’heure de l’action ». Cette exigence pourra ensuite être vérifiée lors des tests de validation.
4. Tester le système (IQ, OQ, PQ)
Pour organiser la validation, des tests vont être réalisés en suivant un plan de test. Celui-ci définit la stratégie de validation, les cas de test à exécuter ainsi que les critères d’acceptation. Ces tests sont répartis en trois phases.
IQ
- IQ (Installation Qualification) : cette étape permet de vérifier que le système a été installé correctement, dans l’environnement prévu et avec les bonnes configurations.
Exemple : vérifier que la version du logiciel installée correspond à celle validée et que tous les composants nécessaires sont correctement configurés.
OQ
- OQ (Operational Qualification) : cette phase consiste à vérifier que le système fonctionne conformément aux exigences définies dans les URS.
Exemple : contrôler qu’un workflow d’approbation suit bien toutes les étapes prévues, que les bonnes personnes sont sollicitées et que chaque action est correctement enregistrée.
PQ
- PQ (Performance Qualification) : cette dernière étape valide que le système répond aux besoins des utilisateurs dans des conditions réelles d’utilisation.
Exemple : demander aux utilisateurs métier de réaliser leurs principaux cas d’usage (création, validation ou signature d’un contrat, par exemple) afin de confirmer que le système répond bien à leurs besoins opérationnels.
5. Documenter les résultats de validation
Durant la phase de validation, des anomalies peuvent apparaitre, il faut les identifier, les tracer et analyser leur impact. Une correction ou justification doit être apportée à chaque test non concluant. À la fin de la validation, un rapport est rédigé. Il résume les tests effectués et leurs résultats, les déviations et résolutions. Ce rapport indique si le système est validé ou non. Dans une validation GxP, la documentation est très importante.
Les principaux référentiels utilisés
Les étapes de la validation sont aujourd’hui un alignement de plusieurs référentiels complémentaires qui définissent la validation GxP. Il s’agit notamment des référentiels suivants :
-
- GAMP 5 : méthodologie de référence fondée sur une approche par les risques pour la validation des systèmes informatisés ;
- EU GMP Annex 11 : exigences réglementaires européennes applicables aux systèmes informatisés utilisés dans les environnements GxP ;
- FDA 21 CFR Part 11 : réglementation américaine relative aux enregistrements électroniques et aux signatures électroniques ;
- ICH Q9 : principes de gestion des risques qualité (Quality Risk Management) ;
- ICH Q10 : modèle de système qualité pharmaceutique couvrant l’ensemble du cycle de vie des produits.
Comment maintenir un système dans un état validé pendant le RUN ?
Pendant la phase de RUN du système, différentes actions doivent être mise en place pour maintenir un état validé via :
- une gestion des changements (change control) ;
- et une gestion des incidents.
Les auditeurs attendent avant tout une démonstration claire que le système reste en permanence dans un état validé et maîtrisé. Pour cela, chaque changement doit être formalisé via un processus de change control rigoureux. Celui-ci inclut une analyse d’impact documentée afin d’évaluer les risques potentiels pour la qualité, la sécurité des patients et l’intégrité des données. Les activités de validation associées doivent être proportionnées au niveau de risque, conformément aux principes GAMP 5. Elles sont systématiquement revues et approuvées par la qualité avant mise en œuvre.
En parallèle, les incidents doivent être gérés de manière structurée et traçable : chaque anomalie doit faire l’objet d’une évaluation de criticité, d’une analyse d’impact (notamment sur les données GxP) et, si nécessaire, de la mise en place d’actions correctives et préventives.
De manière générale, les auditeurs vérifient que toutes les actions sont documentées, justifiées, traçables et auditées, afin de garantir que le système reste fiable, conforme et maîtrisé tout au long de son cycle de vie.
La gestion des changements (Change Control) : cas concrets
-
Dans le cadre de la gestion des changements, lorsqu’une nouvelle version du logiciel est déployée par son éditeur, il est nécessaire d’en analyser les risques afin de déterminer si une nouvelle validation est requise. Cette analyse s’appuie notamment sur un examen détaillé des release notes. Si elle met en évidence un impact sur les exigences GxP, des tests de non-régression (OQ partielle) devront être réalisés avant la mise en production.
Le principal risque est qu’une nouvelle version modifie le comportement du système de manière non maîtrisée, avec des conséquences potentielles sur les processus réglementés en cours.
-
Autre cas fréquent : la modification du profil d’un utilisateur. Ce changement doit suivre un processus formalisé comprenant une demande de modification, une approbation par le manager, puis une mise en œuvre tracée dans le système.
L’objectif est de garantir que chaque utilisateur dispose uniquement des droits d’accès correspondant à son rôle et d’éviter tout accès non autorisé aux données ou aux fonctionnalités du système.
- Dernier exemple en matière de gestion des changements, l’ajout d’un nouveau champ dans un formulaire peut également nécessiter une validation. Une analyse d’impact permet de déterminer si cette nouvelle donnée est critique au regard des exigences GxP. Si c’est le cas, les URS devront être mises à jour, des tests réalisés, puis la modification validée avant sa mise en production. Cette démarche permet de prévenir les risques de corruption, de perte ou d’incohérence des données.
La gestion des incidents
La gestion des incidents a pour objectif de gérer les anomalies en production et garantir la qualité des données.
Par exemple, en cas d’indisponibilité du système, l’incident doit être vite remonté pour éviter toute interruption prolongée d’activité voir une perte de données. On va dès lors activer un plan de continuité, analyser les causes de cette indisponibilité, vérifier l’intégrité des données après redémarrage et faire un rapport de l’incident.
En RUN, il faut donc maintenir l’état validé en contrôlant ce qui change, gérer ce qui ne fonctionne pas et tout documenter. Cette démarche contribue également à favoriser une adoption durable du CLM lorsque celui-ci est utilisé dans un environnement réglementé.
Tous les systèmes informatiques sont-ils concernés par la validation GxP ?
Quels outils peuvent entrer dans le périmètre GxP ?
Contrairement à une idée reçue, la validation GxP ne concerne pas uniquement les logiciels de production. Tout système informatique qui intervient dans un processus réglementé ou qui manipule des données critiques peut entrer dans son périmètre. C’est notamment le cas de certains ERP, des systèmes de gestion documentaire (GED), des logiciels qualité (QMS), des systèmes de laboratoire (LIMS), mais aussi, dans certaines situations, des outils de Contract Lifecycle Management (CLM).
Pourquoi un outil CLM peut-il devenir un système GxP ?
Un outil de Contract Lifecycle Management (CLM) n’est pas, par nature, soumis aux exigences GxP. Tout dépend des contrats qu’il gère et du rôle qu’il joue dans les processus de l’entreprise.
Lorsqu’il intervient dans la gestion de contrats ayant un impact sur la qualité des produits, la conformité réglementaire ou les audits, il peut alors entrer dans le périmètre de la validation GxP.
En effet, un CLM peut être à la fois :
- un simple outil de gestion administrative de contrats ;
- ou un système critique impliqué dans des processus réglementaires.
Tout dépend, encore une fois, de son usage réel. C’est donc un cas concret d’application de la logique vue précédemment : ce n’est pas l’outil qui est GxP, mais l’impact qu’il a sur la qualité, les données et la conformité réglementaire.<
La validation GxP ne consiste pas uniquement à vérifier qu’un logiciel fonctionne correctement. Elle vise à démontrer, de manière documentée, qu’un système informatique reste fiable, sécurisé et conforme tout au long de son cycle de vie. Dans les industries réglementées, cette démarche constitue un levier essentiel pour garantir la qualité des produits, l’intégrité des données et la conformité réglementaire. Elle s’applique à un nombre croissant de systèmes informatisés, dont certains outils de gestion contractuelle lorsque leur usage devient critique.
FAQ – Validation GxP
Qu’est-ce que la validation GxP ?
La validation GxP est une démarche qui permet de démontrer qu’un système informatique utilisé dans un environnement réglementé fonctionne de manière fiable, sécurisée et conforme aux exigences applicables. Elle garantit notamment l’intégrité des données, la traçabilité des actions et la conformité aux réglementations des industries pharmaceutiques, biotechnologiques et des dispositifs médicaux.
Quelle différence entre les GxP et la validation GxP ?
Les GxP regroupent les bonnes pratiques réglementaires applicables aux industries réglementées, comme les GMP (Good Manufacturing Practice – bonnes pratiques de fabrication), GLP (Good Laboratory Practice – bonnes pratiques de laboratoire) ou GCP (Good Clinical Practice – bonnes pratiques cliniques). La validation GxP est la méthode utilisée pour démontrer qu’un système informatique respecte ces exigences lorsqu’il est utilisé dans un processus soumis à ces réglementations.
Quels systèmes informatiques doivent être validés ?
Tous les systèmes informatiques ayant un impact sur la qualité des produits, la sécurité des patients, l’intégrité des données ou la conformité réglementaire peuvent être concernés. Il peut s’agir d’ERP, de systèmes qualité, de logiciels de laboratoire, de GED ou, dans certains cas, d’outils de Contract Lifecycle Management (CLM).
Quelles sont les étapes d’une validation GxP ?
Une validation GxP comprend généralement cinq étapes :
- définir un plan de validation ;
- réaliser une analyse de risques ;
- formaliser les exigences utilisateurs (URS) ;
- exécuter les tests de qualification (IQ, OQ et PQ) ;
- puis documenter l’ensemble des résultats afin de démontrer que le système est conforme.
Quelle différence entre IQ, OQ et PQ ?
L’IQ (Installation Qualification) vérifie que le système est correctement installé.
L’OQ (Operational Qualification) confirme que ses fonctionnalités répondent aux spécifications prévues.
Enfin, la PQ (Performance Qualification) valide que le système fonctionne correctement dans les conditions réelles d’utilisation par les utilisateurs métiers.
Un logiciel SaaS est-il concerné par la validation GxP ?
Oui, un logiciel SaaS peut être soumis à une validation GxP s’il est utilisé dans un processus réglementé et qu’il a un impact sur la qualité, la conformité ou l’intégrité des données. Ce n’est donc pas son mode d’hébergement qui détermine la nécessité de le valider, mais son usage et le niveau de risque associé.
Références principales
- EU GMP Annex 11 – Computerised Systems (EMA)
- 21 CFR Part 11 (FDA – Electronic Records / Electronic Signatures)
- GAMP 5 (ISPE) : A Risk-Based Approach to Compliant GxP Computerized Systems (guide principal pour la validation des systèmes informatisés)

